Protein Isolation
In this post, I will give brief information about Protein Isolation. This can be written in exams and few questions from this topic may appear in competitive exams.
UV absorption at 280 nm is an inaccurate method of protein identification because proteins have different amounts of tyrosine and tryptophan residues, and nucleic acids also absorb at 280 nm.

Isolation of Proteins:
Separation by Molecular Size:
To comprehend the structure and capacity of a protein, one
must know the number and kinds of amino acids present in the protein and their
order. This information is necessary to understand the effects of mutations,
mechanisms of enzyme-catalyzed reactions, and chemical synthesis of
species-specific peptides that may eliminate undesirable hypersensitivity
reactions. Studies of amino acid sequences in proteins have aided in the
understanding of the evolutionary development of living systems. Another
application of amino acid sequence determination is in recombinant DNA
technology. A desired DNA coding for a given polypeptide can be constructed
from knowledge of the precise sequence of that polypeptide.
Bovine insulin (M.W. 5,700) was the first protein to be
completely sequenced by Sanger in 1955. Sanger was also the first to deduce the
base sequence of a DNA molecule obtained from phage ΦX 174. Bovine insulin comprises two peptide chains of 21 and 30
amino acids each, linked by two interchain disulfide bonds. In 1960, Hirs,
Moore, Stein, and Anfinsen explained the first primary structure of the enzyme ribonuclease (M.W. 13,700),
which has a single peptide chain of 124 amino acid residues and four intrachain
disulfide bonds.
Quantitative Determination of Proteins:
UV absorption at 280 nm is an inaccurate method of protein identification because proteins have different amounts of tyrosine and tryptophan residues, and nucleic acids also absorb at 280 nm.
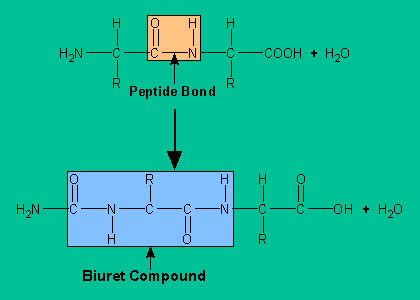
Biuret Test:
In the biuret test, the sample is treated with an
alkaline copper sulfate reagent that produces a violet color and requires a
peptide with at least two peptide bonds. The violet color is produced through
formation of a coordination complex that is analogous to the structure of the complex of biuret with cupric ion.
 |
| Biuret Test |
Folin-Ciocalteu Reaction:
In the Folin-Ciocalteu reaction, the protein reacts with
the phosphomolybdotungstic acid reagent which gives a blue color through reaction with tyrosyl residues. The sensitivity of this
method rely on the amino acid composition of proteins in each sample.
- Protein content, particularly in urine or cerebrospinal fluid, might also be estimated by methods based on precipitation using sulfosalicylic acid.
- The turbidity, which is a measure of protein concentration, can be quantitated by spectrophotometric absorbance methods or light scattering analysis.
- Absorbance of a hydrophobic dye that binds to protein and changes color is also used. Specific proteins in biological fluids can be estimated by more different methods, such as electrophoresis, specific binding techniques, or immunochemical methods.
Determination of Primary Structure of a Protein:
The determination of
the primary structure of a protein consists of the following steps:
1. Obtain a pure protein.
2. Determine the amino acid composition and molecular weight of the pure protein. From amino acid
composition and molecular weight data, calculate the number of residues of each amino acid present per protein molecule to the nearest whole number.
3. Reduce disulfide bonds to sulfhydryl groups.
4. Determine amino terminal (N-terminal) and carboxy terminal (C-terminal) amino acids. A unique residue for each terminus suggests that the native protein contains only one peptide chain.
5. Fragment aliquots of the polypeptide by enzymatic or chemical hydrolysis and separate the peptide mixtures into individual fragments. This process will yield overlapping sets of smaller peptides.
6. Sequence each fragment and, by analyzing the overlapping parts, assemble the sequence of the
original protein.
1. Obtain a pure protein.
2. Determine the amino acid composition and molecular weight of the pure protein. From amino acid
composition and molecular weight data, calculate the number of residues of each amino acid present per protein molecule to the nearest whole number.
3. Reduce disulfide bonds to sulfhydryl groups.
4. Determine amino terminal (N-terminal) and carboxy terminal (C-terminal) amino acids. A unique residue for each terminus suggests that the native protein contains only one peptide chain.
5. Fragment aliquots of the polypeptide by enzymatic or chemical hydrolysis and separate the peptide mixtures into individual fragments. This process will yield overlapping sets of smaller peptides.
6. Sequence each fragment and, by analyzing the overlapping parts, assemble the sequence of the
original protein.
Isolation of Proteins:
- Proteins are separated on the basis of differences in their size, shape, charge, solubility, and binding affinity for other molecules. Most separations begin with proteins in solution. However, when proteins are an integral part of an organelle's structure, it is first necessary to extract them from membranous elements.
- Chemicals used to extract proteins from membranous particles include dissociating agents (urea and mercaptoethanol), chelating agents (ethylenediaminetetraacetic acid), and organic detergents (sodium deoxycholate, sodium lauryl sulfate, and Triton X-100).
- Since the total quantity of a protein in a tissue sample is difficult to identify, the original quantity of protein at the start of a purification is usually based on measurements made on an aliquot of the initial homogenate.
- Each step in a purification process should remove irrelevant protein and retain most of the protein of interest.
- A pure protein preparation is operationally defined as one that maintains a high activity per gram of protein following several purification steps, i.e., the optimum specific activity of the protein. Alternatively, a pure protein cannot be further subdivided by various methods e.g., chromatography or electrophoresis.
- The initial purification steps usually separate proteins according to general classification, e.g., fibrous (insoluble) or globular (soluble).
- The fibrous and globular designations are related to shape and solubility.
- Globular proteins are spherical or ellipsoidal and make up the majority of known proteins.
- Fibrous proteins contain one or more polypeptide chains.
- Their molecules are elongated and asymmetrical with lengths that may be many times their diameters.
- Lateral cross-linking between adjacent polypeptides, by a variety of types of chemical bonding, confers mechanical strength and water insolubility to fibrous proteins; consequently, they are found in connective, elastic, and contractile tissues as well as in hair and skin.
- An initial aqueous extraction procedure tends to partition globular proteins into the soluble fraction and fibrous proteins into the "insoluble pellet" remaining after centrifugation.
Protein can be
separated on the basis of their size by dialysis,
gelfiltration and membrane
filtration.
Ultrafiltration:
Small molecules such as NaCl, amino acids, and sucrose
originally present or added during the separation of organelles can be removed
by dialysis through a semipermeable membrane. Dialysis membranes are prepared
from cellophane or collodion and contain pores that permit passage of solute
molecules whose molecular weight is less than ~5000. Thus, proteins of high
molecular weight are retained within a dialysis bag, whereas low-molecular
weight solutes diffuse through the pores into the fluid (dialysate) outside of
the bag. Complete removal of low molecular-weight solutes requires repeated
changes of dialysate. Rapid removal of low-molecular-weight solutes and
simultaneous concentration of the protein solution can be accomplished by
applying pressure to the dialysis solution (or vacuum to the dialysate). Such a
process is known as ultrafilteration.
Membrane Filtration:
The principles of membrane filtration are the same as those
of dialysis except that synthetic membranes with specified pore sizes are used.
Gel Filtration:
In gel filtration (or molecular sieving), a column is packed with hydrated,
insoluble gel particles with known pore sizes; the protein solution is passed
through the column and the effluent solution is collected in fractions that will
contain solutes of different sizes. The volume of the column is
essentially divided into two phases: the gel phase (within the pores) and the
solvent phase (outside the gel particles). As a solution migrates in the column,
the solute molecules that can penetrate the pores of the gel are distributed
both within the gel and outside it.
The particles that are larger than the pore size are excluded from the gel phase
flow through the column more rapidly and appear first in the effluent. Commonly
used gel particles are inert cross-linked dextrans or agarose, which are commercially
available with a wide range of exclusion limits. Molecular sieving is effective
in the purification of macromolecules and if the column has been calibrated by elution of solutes of known molecular weight, it can also be used in the
estimation of molecular weights of proteins or other solute.
Separation by Chromatography:
Chromatographic separations
of proteins are based upon the differential partitioning of solute molecules
due to their differences in affinity between a moving solvent phase and a fixed
or supportive phase. In gas chromatography, the mobile phase is a gas, whereas
in liquid chromatography it is a liquid. Gas chromatography is not useful in
protein purification because proteins cannot be converted to gases without
decomposition. Liquid chromatography of proteins is performed on a variety of
mechanically different stationary phases, e.g., paper, finely divided particles
coated onto a glass or a plastic surface (thin-layer chromatography), or beads
packed in a column.
Ion Exchange Chromatography:
- Ion exchange chromatography uses an ion exchange resin, and the proteins are eluted with buffer solutions differing in ionic strength and pH.
- The resins are inert polymers to which ionizable groups have been attached; resins with negative charges are cation exchangers, and those with positive charges are anion exchangers.
- Two ion exchange resins frequently used in protein purification are diethylaminoethylcellulose (DEAE-cellulose, an anion exchanger) and carboxymethylcellulose (CM-cellulose, a cation exchanger).
- A protein with a net positive charge at a given pH will combine with the negative groups of a cation exchange resin, and its flow will be retarded; a protein with a net negative charge will migrate through the cation exchange resin unimpeded.
- Cationic proteins in a mixture going through the column will compete with one another for binding to the negatively charged groups of the resin.
- The relative migration rates of different molecules depend on three factors: their individual affinities for the charged sites on the resin, the degree of ionization of the functional groups attached to the resin, and the chemical properties and concentrations of competing low-molecular-weight ions like potassium and sodium.
 |
| Ion exchange Chromatography |
Affinity Chromatography:
- Affinity chromatography takes advantage of specific affinities between protein molecules and analogues of biological molecules that are covalently bound to the column matrix.
- The analogues on the column are usually small molecules resembling enzyme substrates, hormones, or antigens.
- When a protein solution is applied to the column, only those proteins with a high affinity for the matrix are bound.
- Proteins that do not specifically bind pass rapidly through the column.
- The bound proteins can be eluted by altering the pH or ionic strength of the eluent or by adding excess quantities of the ligand, e.g., hormone, antigen, or enzyme substrate or inhibitor.
- Affinity chromatography is useful in the purification of enzymes, hormones and their receptor sites, immunoglobulins and nucleic acids.
 |
| Affinity Chromatography |
Affinity Tag Chromatography:
- Affnity tag chromatography permits purification of recombinant proteins from growth media or from cell lysates.
- New chromatography techniques take advantage of DNA cloning that produces recombinant fusion proteins and allows such proteins to be easily isolated.
- Recombinant proteins can be modified to contain affinity tag sequences to create a fusion protein. The tag possesses unique affinity characteristics that serve as the basis for subsequent isolation. Affinity chromatography is carried out using the immobilized ligand of the tag, which gives a highly purified fusion protein.
- A variety of affinity tag sequences are used such as hexa-histidine for metal chelate separation, enzyme tags that allow isolation using immobilized substrate, or epitope sequences for separation by an immobilized monoclonal antibody.
- An enzyme cleavage site is usually included between the tag and protein for removal of the tag from the fusion protein after isolation.
- Once an effective purification strategy has been established for one fusion protein, it can be used for any protein that is modified to include the same tag.
 |
| Affinity tag Chromatography |
Separation by Electrophoresis:
Electrophoresis separates charged proteins on the basis of their
different mobilities in an electric field. When a solution of proteins is
subjected to an electrical potential, the charged protein molecules migrate
toward either the anode or the cathode. Factors that influence the rate of
migration are pH, composition of the medium, through which migration occurs,
and size and shape of the protein molecule.
Isoelectric Focusing:
- A protein that does not migrate in an electric field at a given pH has no net charge at that pH. That pH is called the isoelectricpoint (pI) of the protein.
- The pI value is characteristic for each protein. In a solution at a pH value above its pI, a protein will have a net negative charge; below its pI, a protein will have a net positive charge.
- The electrophoresis technique known as isoelectricfocusing separates proteins on the basis of differences in their isoelectric points.
- Proteins whose pI values differ by as little as 0.02 pH unit can be separated by this technique.
- In isoelectric focusing, a mixture of proteins is placed in a pH gradient; in the presence of an electric field, each protein migrates to a position corresponding to its pI and comes to rest in a narrow band at that pH.
- The pH gradient is established by placing in an electric field an aqueous mixture of synthetic low-molecular-weight (300-600) ampholytes.
- On application of an electric potential, the ampholytes migrate and come to rest according to their respective isoelectric points.
- If proteins are mixed with the ampholytes, the proteins migrate to the positions of their respective isoelectric points within the ampholyte gradient and can thereby be separated and concentrated into narrow bands.
 |
| Isoelectric Focusing |
SDS-PAGE:
- Electrophoretic techniques also yield estimates of the molecular weights of proteins and nucleic acids.
- The detergent sodium dodecyl sulfate (SDS) and proteins form SDS-protein complexes that migrate in polyacrylamide gels according to their molecular weights.
- SDS dissociates multi subunit proteins into individual polypeptide chains.
- Each denatured chain has a uniform negative charge per unit mass of protein, since the total negative charge of the sulfonic acid groups of SDS, which are uniformly located along the surface of the protein, far exceeds the intrinsic charge of the amino acid residues.
- SDS-protein complexes, all of which are rod-shaped, migrate toward the anode rapidly if they are small, but slowly if they are large.
- The polyacrylamide medium retards migration according to the size of the rigid rods.
- The molecular weight of a protein is established by comparing its rate of migration with those of a number of proteins of known molecular weight.
 |
| SDS-PAGE |
Capillary Electrophoresis:
- This method is capable of distinguishing between proteins that differ only slightly in amino acid composition or glycosylation.
- In capillary electrophoresis, protein samples are first injected onto a fused silica microcolumn.
- The free Si-O-H groups of the fused silica become ionized at pH values greater than 2, causing the inside surface of the column to be negatively charged; the charge density depends on pH.
- The micro-column sustains electric fields that are at least 50-fold more powerful than those used in slab gel electrophoresis.
- As a result of the high-intensity electric field, isolation of proteins is more readily accomplished and the increased sensitivity enables micro characterization of proteins.
- Due to the small size of the column, nano liter quantities can be analyzed and isolation occurs within minutes, whereas in slab gels it may take several hours.
- As the samples get off the column, proteins are measured by a specialized detector.
 |
| Capillary Electrophoresis |
Separation by Solubility:
- Proteins may also be isolated by selective precipitation, which is usually done by changing the salt concentration (ionic strength) of the solution.
- Many proteins at low salt concentration, that are readily soluble in aqueous solutions exhibit decreased solubility with increased salt concentration.
- Protein precipitation by increasing salt concentration is known as the salting-out technique. In this technique, the salt ions compete with the protein molecules for interaction with water molecules, such that the affinity between protein molecules increases as water molecules are removed from the surface functional groups of the proteins.
- Eventually, the decreased polymer-solvent association causes in precipitation of the protein. Since different proteins possess different surface functional groups, proteins can be differentially precipitated at different salt concentrations.
- Salting-in phenomenon increases the solubility of some proteins by adding dilute salt solution, is believed to be due to interactions between salt ions and the charged groups on protein molecules, which reduce protein-protein interaction and increase protein-solvent interaction.
- Ammonium sulfate is the most regularly used compound for salting out of proteins because it is very soluble (706 g/L) and has four ionic charges per molecule.
- Precipitations are generally performed slowly with cold solutions to minimize protein denaturation due to the heat released on stirring and to allow time for the formation of precipitates.



Comments
Post a Comment